


Le amiloidosi sono un gruppo di malattie rare, endocrine e metaboliche, piuttosto vario, causate dalla capacità di alcune proteine prodotte dal nostro organismo di cambiare la loro conformazione. Facendo questo, diventano in grado di formare sia strutture solubili tossiche, dette oligomeri, sia complessi di fibre insolubili che possono depositarsi in vari tessuti e/o organi, compromettendone la funzionalità.
Gli aggregati fibrillari vengono definiti anche depositi amiloidogenici, termine che deriva dal latino amiloideus usato per la prima volta nel 1854 da Rudolf Virchow per indicare un piccolo deposito di natura proteica dalle proprietà simili all’amido.
Ad oggi, sono state identificate 42 proteine amiloidogeniche ed ognuna di esse è associata ad una specifica forma di amiloidosi.
Le amiloidosi più diffuse sono quelle centrali, come l’Alzheimer e il Parkinson, caratterizzate da depositi fibrillari a livello del sistema nervoso centrale.
Meno diffuse sono invece le amiloidosi sistemiche, in cui le proteine amiloidogeniche trasportate dal sangue possono raggiungere vari organi, quali il fegato, il cuore o i reni, in cui si depositano e formano gli aggregati fibrillari. Ad oggi sono state individuate circa 20 proteine che nell’uomo sono responsabili di amiloidosi sistemiche, come:
Le amiloidosi possono essere divise anche in:
L’amiloidosi da catene leggere delle immunoglobuline, chiamata anche Amiloidosi AL, è la forma più comune di amiloidosi sistemica nei paesi occidentali, con un’incidenza di circa 10 nuovi casi su un milione di persone all’anno.
È causata da un'alterazione delle caratteristiche chimico-fisiche di una popolazione di plasmacellule geneticamente identiche (insieme tecnicamente definito clone).
Queste cellule sono quelle che, nel midollo osseo, normalmente producono e assemblano le immunoglobuline, più comunemente conosciute col nome di anticorpi.
Le immunoglobuline sono formate da un complesso a forma di “Y” costituito da due catene leggere (Light Chain) identiche legate a due catene pesanti.
Le catene leggere sono fisiologicamente prodotte nel midollo osseo in lieve eccesso rispetto alle catene pesanti. La maggior parte di esse si lega alle catene pesanti, mentre l’eccesso viene rilasciato nel flusso ematico raggiungendo i reni. Qui, grazie alle loro piccole dimensioni, le LC vengono eliminate.
Nei pazienti affetti da amiloidosi AL il clone anomalo di plasmacellule produce una quantità eccessiva di catene leggere, più di quante l’organismo riesca a smaltire, con un conseguente loro aumento nel sangue. Grazie alla loro capacità di auto-aggregarsi, le LC si depositano in vari organi (ad esclusione del cervello), ostacolandone la normale funzione e causando un danno specifico.
E’ stato osservato che circa il 75% dei pazienti affetti da amiloidosi AL presenta depositi a livello cardiaco e il 65% a livello renale. Il danno che ne consegue può essere di tipo funzionale, con un’alterazione del funzionamento di tutti i processi fisiologici del tessuto coinvolto, o strutturale, accompagnato cioè da un difetto nell’architettura degli organi.
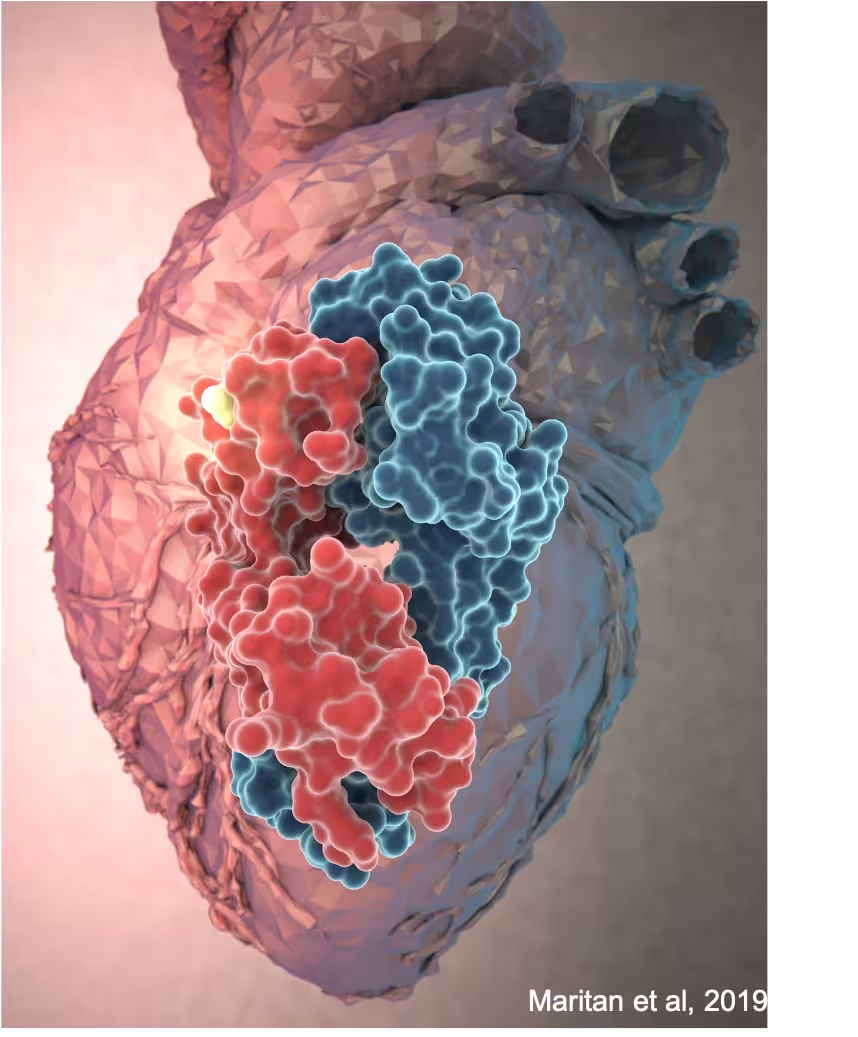
I pazienti affetti da amiloidosi AL con depositi a livello cardiaco sono quelli con la prognosi peggiore: se non sottoposti tempestivamente alle cure, la loro sopravvivenza varia dai 6 ai 15 mesi dalla diagnosi.
Anche nei pazienti in cui i depositi di LC si localizzano in organi diversi dal cuore, è stato visto che la principale causa di morte è rappresentata dall’insorgenza di una grave insufficienza cardiaca e di alterazioni nel normale ritmo di contrazioni del cuore.
L’amiloidosi da catene leggere è caratterizzata da una varietà di sintomi, per lo più aspecifici, che possono rendere difficile la diagnosi.
I sintomi precoci sono infatti molto generici e possono essere comuni a molte patologie:
Generalmente la diagnosi viene effettuata alla comparsa dei sintomi più gravi a carico dell’organo in cui si sono depositate le LC. Questi danni spesso sono irreversibili.
A seconda di quale sia l’organo colpito dalla patologia, la malattia si manifesta in maniera differente in ciascun paziente.
I sintomi di questa fase, che possono comparire singolarmente o in concomitanza l’uno dell’altro, comprendono:
La difficoltà diagnostica di questa patologia deriva dal fatto che i sintomi sono variabili, aspecifici e incredibilmente tardivi nella loro comparsa.
Esistono tuttavia dei campanelli d’allarme. Infatti, un coinvolgimento dello stesso clone di plasmacellule è evidente nei pazienti già 4 anni prima dell’insorgenza dei primi sintomi. In aggiunta, esistono dei biomarcatori, dosabili nel sangue o nelle urine, i cui livelli si modificano ben prima della comparsa dei sintomi. I più importanti sono il NT-proBNP e l’albuminuria, rilevabili con un semplice test del sangue per valutare rispettivamente la funzionalità cardiaca e quella dei reni.
È importante inoltre valutare la comparsa nel sangue di un eccesso di LC, ovvero di una popolazione monoclonale anomala.
Indagini cliniche successive comprendono una biopsia dei tessuti nei quali si sospetta la presenza di depositi di catene leggere.
E’ poi consigliato effettuare in parallelo dei test genetici per distinguere le forme sporadiche di amiloidosi da quelle ereditarie.
Esistono alcune condizioni che espongono una persona ad un maggior rischio di sviluppare l'amiloidosi da catene leggere, come:
L’obiettivo terapeutico principale nella gestione di questa patologia è quello di ridurre i livelli di catene leggere circolanti mediante l’eliminazione del clone plasmacellulare anomalo.
A questo proposito vengono attualmente utilizzati dei cocktail di chemioterapici, chiamati bortezomib, associati a cortisone. Di frequente questo trattamento si associa all’auto-trapianto di cellule staminali, chiamato anche trapianto autologo. I pazienti idonei al trapianto autologo dispongono della strategia terapeutica più efficace, con comprovati miglioramenti della funzionalità cardiaca.
Sebbene quasi il 70% dei pazienti risponda in modo positivo alle cure, esiste una piccola parte di pazienti che arriva alla diagnosi con il cuore ormai già molto compromesso. Purtroppo, questi pazienti non sono in grado di sopportare una terapia così aggressiva. Quindi, allo stato attuale, lo sviluppo di una terapia efficace anche per questa fascia di pazienti resta la necessità più urgente.
L’amiloidosi AL, come spesso accade per le malattie rare, non riesce a beneficiare di grandi investimenti economici per lo studio e per lo sviluppo di nuove terapie farmacologiche. Nonostante gli sforzi degli ultimi 20 anni da parte di diversi gruppi di ricerca, non esiste purtroppo ancora un modello animale in grado di riprodurre le caratteristiche principali della patologia.
L’unico modello finora sviluppato e utilizzato con successo è quello che utilizza il nematode Caenorhabditis elegans. Il Laboratorio di Patologia Umana in Organismi Modello studia i meccanismi molecolari alla base dell’aggregazione delle proteine amiloidogeniche, compresa l’amiloidosi AL, in questo organismo modello sperimentale.

Inoltre, essendo un animale invertebrato, il suo utilizzo non pone i problemi di natura etica comunemente legati alla sperimentazione animale.
I ricercatori guidati dalla Dr.ssa Luisa Diomede hanno dimostrato che le catene leggere amiloidogeniche generano alti livelli di specie reattive dell’ossigeno o radicali liberi, dannosi per il nostro organismo. L’aumento di radicali liberi provoca un danno tissutale e funzionale della faringe del nematode, che può essere considerata un “cuore ancestrale”. Questo effetto è strettamente dipendente dalla presenza di ioni metallici, in particolare del rame.
La somministrazione di farmaci in grado di ripristinare la giusta concentrazione dei metalli blocca la produzione di radicali e previene l’effetto tossico delle LC. Sulla base di questi risultati è stato disegnato, in collaborazione con il Centro per la Cura delle Amiloidosi del Policlinico San Matteo di Pavia, uno studio clinico di fase II su pazienti AL con coinvolgimento cardiaco.
Luisa Diomede e Oscar Fumagalli - Laboratorio di Patologia Umana in Organismi Modello
Editing Raffaella Gatta - Content manager

Intercheck web: applicazione web rivolta a medici e operatori sanitari a supporto della corretta prescrizione dei farmaci nell’anziano.

Servizio di informazioni rivolto alle mamme in gravidanza e allattamento sul corretto uso dei farmaci.


Servizio di informazioni rivolto a medici, operatori sanitari e cittadini sui piani terapeutici e le patologie rare.

Servizio di informazioni rivolto ai medici, agli operatori sanitari e ai pazienti sulle malattie renali e i trapianti.
Servizio di informazioni rivolto a medici, operatori sanitari e cittadini sull'uso corretto dei farmaci, sugli effetti collaterali e le interazioni nell'anziano.
Intercheck web: web application aimed at health professionals to support the proper prescription of drugs in the elderly.
Information service for pregnant and nursing mothers on the appropriate use of medicines.
Information service for health professionals and patients on treatment plans and rare diseases.
Information service for health professionals and patients on kidney diseases and transplants.
Information services aimed at health professionals and the general public on the correct use of drugs, side effects and interactions in the elderly.
